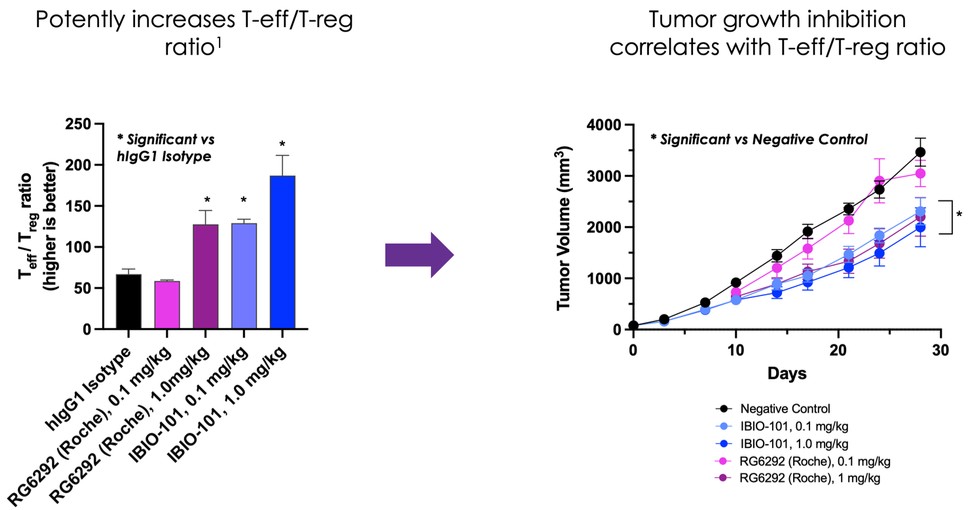
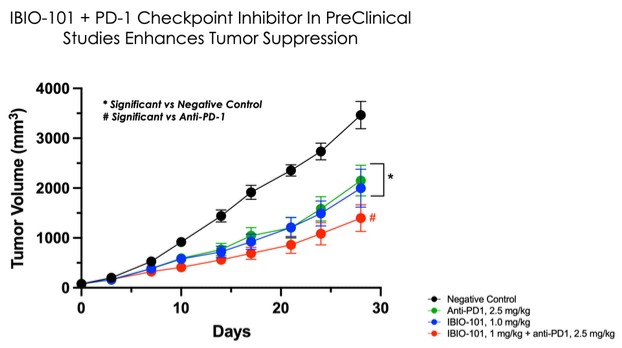
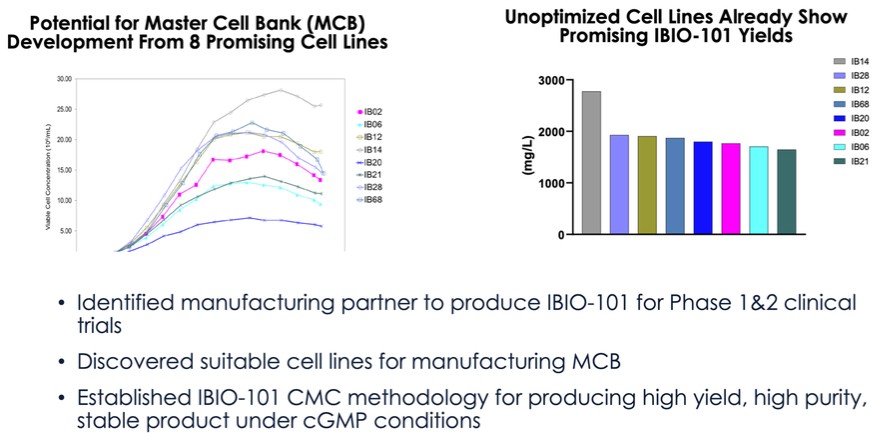
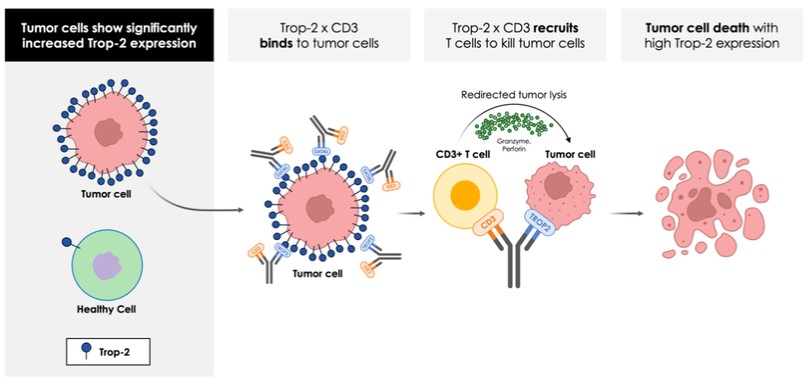
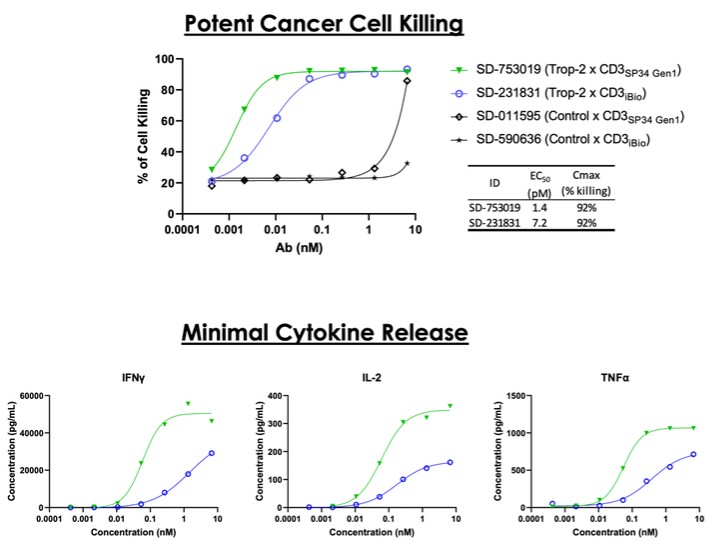
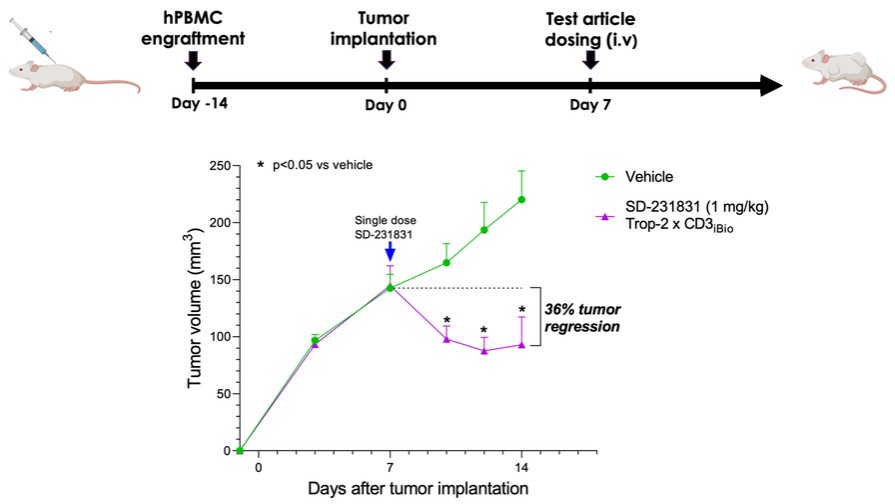
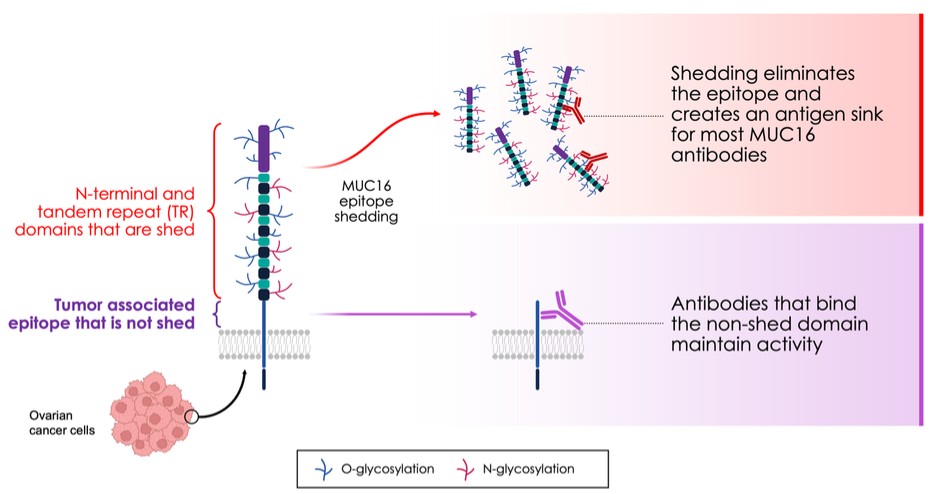
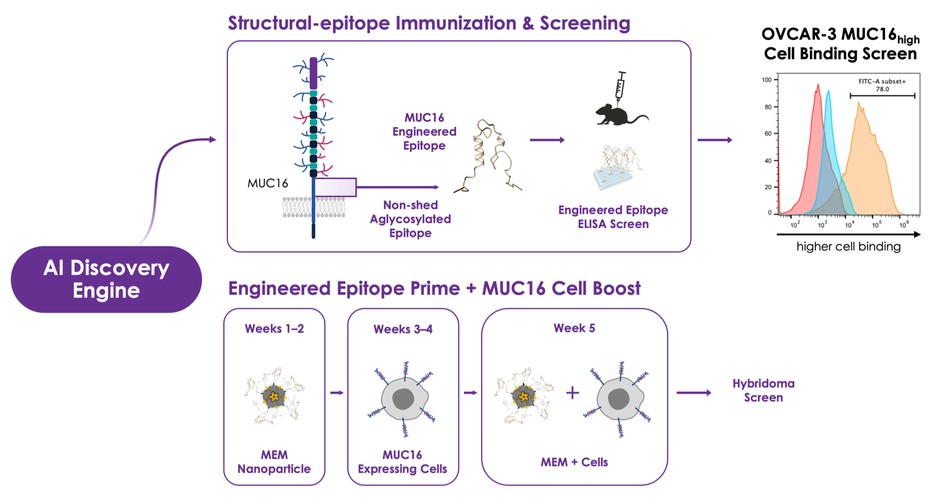
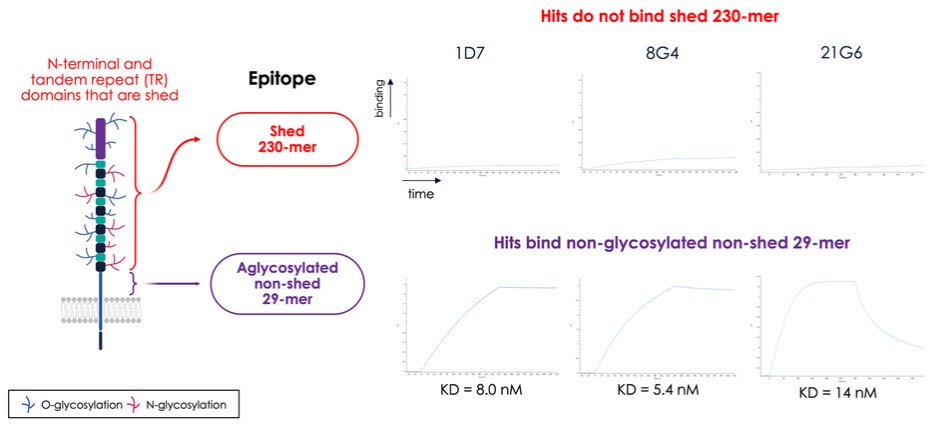
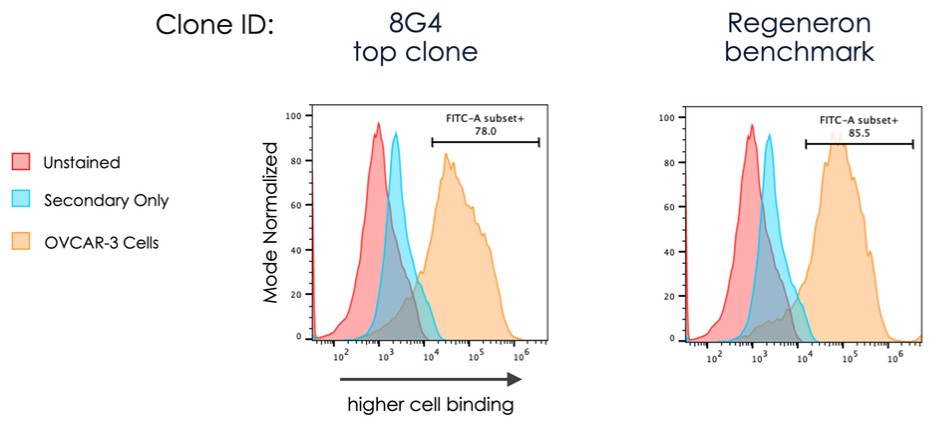
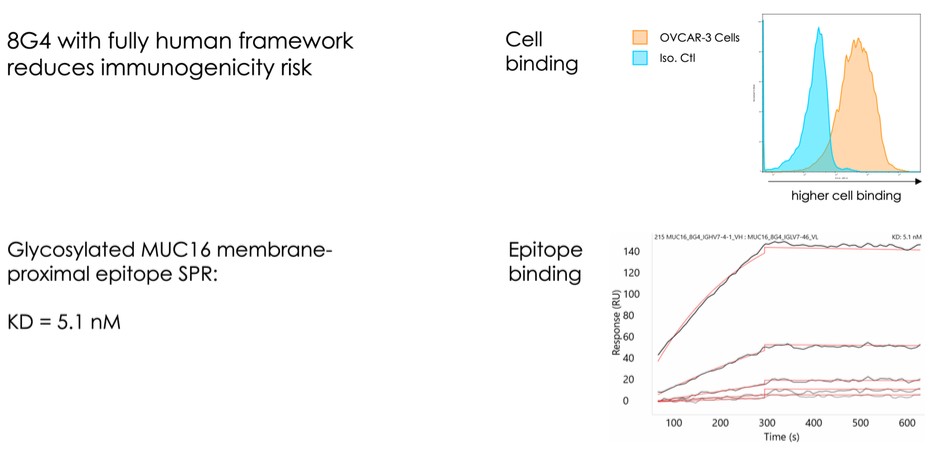
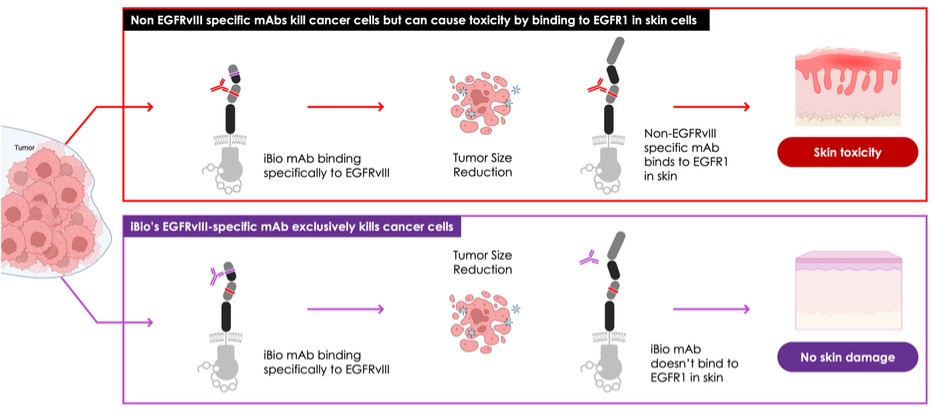
dependent upon a specified milestone. In addition, the Company agreed that each time it consummated an at-the-market issuance of Equity Interests (as defined within the Credit Agreement), no later than five (5) days following such issuance of Equity Interests, it would (i) pay to Woodforest in immediately available cash funds, without setoff or counterclaim of any kind, forty percent (40%) of the Net Proceeds (as defined within the Credit Agreement) received by the Company for such issuance of Equity Interests; provided, any such payment would cease upon payment obligations in full and (ii) provide Woodforest with a detailed accounting of each such issuance of Equity Interests.
On March 24, 2023, iBio CDMO and Woodforest entered into a fourth amendment to the Credit Agreement (the “Fourth Amendment”), which within the Fourth Amendment Woodforest agreed to (i) reduce the percentage of any payment to Woodforest the Company was required to make from the proceeds of sales of its common stock under its at-the-market facility from 40% to 20%, (ii) reduce the percentage of any payment to Woodforest the Company was required to make from the proceeds of sales of its equipment from 40% to 20%, and (iii) allowed the Company to retain $2,000,000 million of the $5,100,000 million that the Company received from the Fraunhofer Settlement Funds, with the remaining $3,000,000 million being held in a Company account at Woodforest. In addition, the Company was obligated to (y) deliver to Woodforest an executed copy of a purchase agreement (the “Woodforest Purchase Agreement”) for the sale of the Facility, no later than April 14, 2023, and (z) pay to Woodforest a fee in the amount of $75,000 on the earlier of the date of the closing of the Woodforest Purchase Agreement, or the maturity date of the term loan (the “Maturity Date”). In addition, on March 24, 2023, the Company, as guarantor, entered into a fourth amendment to the Guaranty, which reduced the Liquidity Covenant from $7,500,000 to $1,000,000.
On May 10, 2023, iBio CDMO and Woodforest entered into a fifth amendment to the Credit Agreement (the “Fifth Amendment”), pursuant to which Woodforest agreed to: (i) waive the obligation to deliver to Woodforest an executed copy of a Woodforest Purchase Agreement for the sale of the Facility no later than April 14, 2023 and, (ii) release $500,000 of the $3.0 million being held in a Company account at Woodforest when the outstanding principal amount was reduced to $10.0 million and for each additional $2.5 million reduction of the outstanding principal amount, an additional $750,000 was to be released from the Company account at Woodforest. In addition, starting on the effective date of the Fifth Amendment, the interest on the Term Loan was increased to 5.25%, and the Term Loan further accrued interest, payable in kind and added to the balance of the outstanding principal amount at a fixed rate per annum equal to (a) 1.00%, if the Facility was sold on or before June 30, 2023, (b) 2.00% if the Facility was sold after June 2023, but on or before September 30, 2023, or (c) 3:00%, if the Facility was sold after September 30, 2023, or not sold prior to the Maturity Date. The Company also agreed to pay Woodforest a fee in the amount of (x) $75,000 if the Facility was sold on or before June 30, 2023, (y) $100,000 if the Facility was sold after June 2023, but on or before September 30, 2023, or (x) $125,000, if the Facility was sold after September 30, 2023, or not sold prior to the Maturity Date.
On September 18, 2023, iBio CDMO and Woodforest entered into a sixth amendment to the Credit Agreement (the “Sixth Amendment”), which amended the Credit Agreement to: (i) set the Maturity Date to the earlier of (a) December 31, 2023, or (b) the acceleration of maturity of the term loan in accordance with the Credit Agreement, (ii) provided that iBio CDMO would, immediately upon receipt of the proceeds of the sale of the Facility, apply the net proceeds to satisfy all outstanding obligations under the term loan, and to the extent such net proceeds were sufficient, to pay off the term loan, and (iii) change the annual filing requirement solely for the fiscal year ending June 30, 2023; provided that (y) iBio CDMO deliver an executed copy of the Purchase and Sale Agreement for the sale of the Facility within one business day after entry into the Sixth Amendment, and (z) if the Facility was not sold on or before December 1, 2023, iBio CDMO would pay a fee in the amount of $20,000 upon the earlier of the date of the closing or the Maturity Date.
On October 4, 2023, iBio CDMO and Woodforest entered into the seventh amendment to the Credit Agreement (the “Seventh Amendment”), which amendment among other things, permitted the Company, in each case, so long as no Potential Default or Default (as such terms were defined in the Credit Agreement) to make the following withdrawals from the Reserve Funds Deposit Account (as defined in the Credit Agreement): (i) up to $1,000,000 on October 4, 2023 so long as iBio CDMO maintained a minimum balance of $2,000,000 until October 16, 2023, (ii) up to an additional $750,000 after October 16, 2023 so long as iBio CDMO maintained a minimum balance of $1,250,000 until November 13, 2023, and (iii) up to an additional $250,000 after November 13, 2023 so long as iBio CDMO maintained a minimum balance of $1,000,000 until Payment in Full (as defined in the Credit Agreement). On the earlier of (a) the closing of the Woodforest Purchase Agreement (as defined in the Credit Agreement), or (b) the Maturity Date, the Company would pay Woodforest $20,000. In addition, on October 4, 2023, the Company, as guarantor, entered into the Fifth Amendment to the Guaranty,